Research
The research conducted in our group is data-driven. That means instead of making theoretical assumptions about a given problem, we are using data generated by experiments to formulate hypotheses and for extracting information and knowledge about the underlying problem. For this reason our research is within the field of data science.
The data we are using for your studies come from a number of different fields, including biology, medicine, economy, finance, industry, social media and social sciences. However, a common denominator to all these different problems is that we are applying statistical thinking in a systematic way in the form of machine learning, artificial intelligence and statistics. This allows us to ensure that the obtained results are robust. A further commonality is that the underlying system variables are heterogeneously connected in the form of networks. This requires in addition network thinking about problems to achieve optimal interpretations.
General Research Interests
Our general research interest is in data science, including the following fields:
-
Computational Biology
-
Network Science
-
Machine Learning/Artificial Intelligence
-
Text mining
Software
Our group developed a number of software solutions for the statistical analysis of data and the visualization of networks. The following software packages have been developed for the programming language R.
-
BC3Net Inference of causal networks (from CRAN)
-
NetBioV Network visualization (from Bioconductor)
-
samExploreR Preprocessing for RNA-seq data (from Bioconductor)
-
mvgraphnorm Generate constrained covariance matrices for Gaussian graphical models (from CRAN)
-
sgnesR Simulating gene expression data from an underlying gene network structure from (GitHub)
-
GSAR Gene Set Analysis in R (from Bioconductor)
Web portal
In addition to software packages we also develop web portals. A web portal allows the interactive exploration of complex and high-dimensional results.
-
Drug association network Network visualization of significant similarities of drugs
-
L1000 Viewer Access to the LINCS L1000 data repository
Our philosophy
We love data
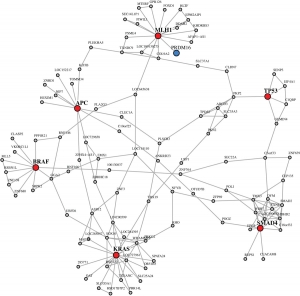
Network Inference
The inference of gene regulatory networks, which is sometimes also referred to as reverse engineering is the process of estimating the direct physical (biochemical) interactions of a cellular system from data. That means one aims for identifying all molecular regulatory interactions among genes that are present in an organism to establish and maintain all required biological functions characterizing a certain physiological state of a cell. Depending on the data used for inferring the network, which, principally, may either come from DNA microarray, RNA-seq, proteomics or ChIP-chip experiments, or combinations thereof, the biological interpretation of an ‘edge’ in these networks is dependent thereon. For gene expression data, inferred interactions may preferably indicate transcription regulation, but can also correspond to protein-protein interactions. Due to the causal character of these networks, which ensures a meaningful biological interpretation, the genome-wide inference of gene regulatory networks holds great promise in enhancing the understanding of normal cell physiology, and also complex pathological phenotypes.
Based on our experience in the development of statistical network inference methods over the last years we developed the methods C3Net and B3Net. Currently, we are planing to extend these methods by using transfer learning.
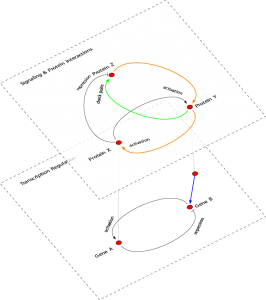
Relation detection with text mining
During the last years there has been a systematic shifted of the focus of attention from the identification of the differential expression of single genes to groups of genes. For this reason, we are developing methods that make explicitly use of correlation structures among genes in a pathway or biological process. The major purpose of this project is to develop multivariate statistical methods for the analysis of microarray data and RNA-seq data from complex diseases to detect pathological pathways to improve existing methods significantly. The importance of the proposed project comes from the fact that it is goal directed towards a functional understanding of cancer on a pathway level. This implies that results obtained by the application of the proposed method have an immediate effect and impact on biomedical and medical research.
We are currently working on the extension of such methods so they can also be used with RNA-seq data from next-generation sequencing technologies. The problem with RNA-seq data is that they are ‘count data’ that makes the immediate translation of most methods impossible, but specific attention needs to be placed on the statistical estimators that are optimizes for such data characteristics. The resulting methodology will be applicable to problems from Biology and the Biomedical Science alike allowing to identify aberrant biological process between physiological conditions.
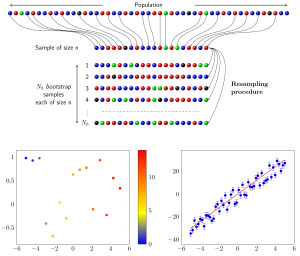
Computational Statistical Methods
The Department of Signal Processing has an excellent reputation for developing statistical methodology, e.g., exemplified by the seminal work of Jorma Rissanen inventing the Minimum Description Length (MDL). We develop statistical methodology with a particular focus on approaches that can be used for the analysis of genomics data. Despite considerable differences between different data types from genomics there are also commonalities. For instance, data of any kind possess a complex correlation structure. This correlation structure is the source of the concerted interaction of genes and gene products manifesting in various molecular networks, e.g., the protein interaction network. Using this phenomenon as a starting point there are a number of related approaches that can be pursued. For pathway/module methods improved estimators for covariance matrices are needed that can better cope with moderate sample sizes. Here ‘moderate’ means that the sample sizes allow to perform resampling approaches, e.g., Boostrap, that enable the combination with ensemble methods. This would allow to replace theoretical assumptions with numerical estimations based on data to enhance, e.g., the power of a hypothesis test. These studies are also related to Graphical Models.
Due to the fact that more and more large-scale high-throughput data are generated, we are increasingly in a position to convert such data into structured networks. However, the statistical comparison of such networks is a difficult problem that requires further attention and problem specific approaches. Based on our experience, we work on entropy-based methods, as a flexible means to emphasize structural and ontological features. We will especially explore the latter aspect to develop biology and disease specific comparisons. Furthermore, we work on general network analysis and graph theory based methods in combination with their computational realization for the partitioning of networks also called module finding, because the resulting modules may enhance our understanding of biological processes and their hierarchical organization structure.